Реакции замещения лигандов. Реакции замещения лигандов в порфириновых комплексах циркония, гафния, молибдена и вольфрама моторина елена викторовна. Вопросы для самоконтроля
Введение к работе
Актуальность работы . Комплексы порфиринов с металлами в высоких степенях окисления могут координировать основания гораздо более эффективно, чем комплексы М 2+ и образовывать смешанные координационные соединения, у которых в первой координационной сфере центрального атома металла наряду с макроциклическим лигандом находятся нециклические ацидолиганды, а иногда и координированные молекулы. Вопросы совместимости лигандов в таких комплексах чрезвычайно важны, так как именно в виде смешанных комплексов порфирины осуществляют свои биологические функции. Кроме того, реакции обратимого присоединения (переноса) молекул оснований, характеризующиеся умеренно высокими константами равновесия, могут успешно применяться для разделения смесей органических изомеров, для количественного анализа, для целей экологии и медицины. Поэтому исследования количественных характеристик и стехиометрии равновесий дополнительной координации на металлопорфиринах (МР) и замещения простых лигандов в них полезно не только с точки зрения теоретических познаний свойств металлопорфиринов как соединений комплексных, но и для решения практической задачи поиска рецепторов и переносчиков малых молекул или ионов. До настоящего времени систематические исследования для комплексов высокозарядных ионов металлов практически отсутствуют.
Цель работы . Настоящая работа посвящена изучению реакций смешанных порфирин-содержащих комплексов высокозарядных катионов металлов Zr IV , Hf IV , Mo V и W V с биоактивными N-основаниями: имидазолом (Im), пиридином (Py), пиразином (Pyz), бензимидазолом (BzIm), характеристике устойчивости и оптических свойств молекулярных комплексов, обоснованию ступенчатых механизмов реакций.
Научная новизна . Методами модифицированного спектрофотометрического титрования, химической кинетики, электронной и колебательной абсорбционной и 1 Н ЯМР спектроскопии впервые получены термодинамические характеристики и обоснованы стехиометрические механизмы реакций N-оснований с металлопорфиринами со смешанной координационной сферой (Х) n-2 МТРР (Х – ацидолиганд Cl - , OH - , O 2- , ТРР - дианион тетрафенилпорфирина). Установлено, что в подавляющем большинстве случаев процессы образования супрамолекул металлопорфирин – основание протекают ступенчато и включает несколько обратимых и медленных необратимых элементарных реакций координации молекул оснований и замещения ацидолигандов. Для каждой из стадий ступенчатых реакций определены стехиометрия, константы равновесий или скорости, порядки медленных реакций по основанию, спектрально охарактеризованы продукты (УФ, видимые спектры для промежуточных продуктов и УФ, видимых и ИК – для конечных). Впервые получены корреляционные уравнения, позволяющие прогнозировать устойчивость супрамолекулярных комплексов с другими основаниями. Уравнения использованы в работе для обсуждения детального механизма замещения ОН - в комплексах Mo и W на молекулу основания. Описаны свойства МР, обуславливающие перспективу использования для детектирования, разделения и количественного анализа биологически активных оснований, такие как умеренно высокая устойчивость супрамолекулярных комплексов, четкий и быстрый оптический отклик, низкий порог чувствительности, секундное время обращения.
Практическая значимость работы . Количественные результаты и обоснование стехиометрических механизмов реакций молекулярного комплексообразования имеют существенное значение для координационной химии макрогетероциклических лигандов. В диссертационной работе показано, что смешанные порфирин-содержащие комплексы проявляют высокую чувствительность и селективность в отношении биоактивных органических оснований, в течение нескольких секунд или минут дают оптический отклик, пригодный для практического детектирования реакций с основаниями - VOCs, компонентами лекарств и пищевых продуктов, благодаря чему рекомендованы для использования в качестве компонентов сенсоров оснований в экологии, пищевой промышленности, медицине и сельском хозяйстве.
Апробация работы . Результаты работы докладывались и обсуждались на:
IX Международной конференции по проблемам сольватации и комплексообразования в растворах, Плес, 2004; XII Симпозиуме по межмолекулярному взаимодействию и конформациям молекул, Пущино, 2004; XXV, XXVI и XXIX научных сессиях Российского семинара по химии порфиринов и их аналогов, Иваново, 2004 и 2006; VI Школе-конференции молодых ученых стран СНГ по химии порфиринов и родственных соединений, Санкт-Петербург, 2005; VIII научной школе - конференции по органической химии, Казань, 2005; Всероссийской научной конференции «Природные макроциклические соединения и их синтетические аналоги», Сыктывкар, 2007; XVI Международной конференции по химической термодинамике в России, Суздаль, 2007; XXIII Международной Чугаевской конференции по координационной химии, Одесса, 2007; International Conference on Porphyrins and Phtalocyanines ISPP-5, 2008; 38th International Conference on Coordination Chemistry, Israel, 2008.
Основная реакция замещения в водных растворах - обмен молекул воды (22) - была изучена для большого числа ионов металлов (рис. 34). Обмен молекул воды координационной сферы иона металла с основной массой молекул воды, присутствующей в качестве растворителя, для большинства металлов протекает очень быстро, и поэтому скорость такой реакции удалось изучить главным образом методом релаксации. Метод заключается в нарушении равновесия системы, например резким повышением температуры. При новых условиях (более высокой температуре) система уже не будет находиться в равновесии. Затем измеряют скорость установления равновесия. Если можно изменить температуру раствора в течение 10 -8 сек , то можно измерить скорость реакции, которая требует для своего завершения промежутка времени больше чем 10 -8 сек .
Можно измерить также скорость замещения координированных молекул воды у различных ионов металлов лигандами SO 2- 4 , S 2 O 3 2- , EDTA и др. (26). Скорость такой реакции
зависит от концентрации гидратированного иона металла и не зависит от концентрации входящего лиганда, что позволяет использовать для описания скорости этих систем уравнение первого порядка (27). Во многих случаях скорость реакции (27) для данного иона металла не зависит от природы входящего лиганда (L), будь то молекулы H 2 O или ионы SO 4 2- , S 2 O 3 2- или EDTA.
Это наблюдение, а также тот факт, что в уравнение скорости этого процесса не включена концентрация входящего лиганда, позволяют предполагать, что эти реакции протекают по механизму, в котором медленная стадия заключается в разрыве связи между ионом металла и водой. Получающееся соединение, вероятно, затем быстро координирует находящиеся поблизости лиганды.
В разд. 4 данной главы было указано, что более высокозаряженные гидратированные ионы металла, такие, как Al 3+ и Sc 3+ , обменивают молекулы воды медленнее, чем ионы M 2+ и M + ; это дает основание предполагать, что в стадии, определяющей скорость всего процесса, важную роль играет разрыв связей. Выводы, полученные в этих исследованиях, не окончательны, но они дают основание думать, что в реакциях замещения гидратированных ионов металлов важное значение имеют S N 1-процессы.
Вероятно, самыми изученными комплексными соединениями являются аммины кобальта(III). Их устойчивость, легкость приготовления и медленно текущие с ними реакции делают их особенно удобными для кинетического изучения. Так как исследования этих комплексов были проведены исключительно в водных растворах, вначале следует рассмотреть реакции этих комплексов с молекулами растворителя - воды. Было установлено, что вообще молекулы аммиака или аминов, координированные ионом Co(III), настолько медленно замещаются молекулами воды, что обычно рассматривают замещение иных лигандов, а не аминов.
Была изучена скорость реакций типа (28) и найдено, что она первого порядка относительно комплекса кобальта (X - один из множества возможных анионов).
Так как в водных растворах концентрация H 2 O всегда равна примерно 55,5 М , то нельзя определить влияние изменения концентрации молекул воды на скорость реакции. Уравнения скорости (29) и (30) для водного раствора экспериментально не различимы, так как к просто равно k" = k". Следовательно, по уравнению скорости реакции нельзя сказать, будет ли H 2 O участвовать в стадии, определяющей скорость процесса. Ответ на вопрос, протекает ли эта реакция по механизму S N 2 с заменой иона X на молекулу H 2 O или по механизму S N 1, предусматривающему вначале диссоциацию с последующим присоединением молекулы H 2 O, нужно получить при помощи других экспериментальных данных.
Решения этой задачи можно добиться двумя типами экспериментов. Скорость гидролиза (замещение одного иона Cl - на молекулу воды) транс - + примерно в 10 3 раз больше скорости гидролиза 2+ . Увеличение заряда комплекса приводит к усилению связей металл - лиганд, а следовательно, и к торможению разрыва этих связей. Следует также учитывать притяжение входящих лигандов и облегчение протекания реакции замещения. Так как обнаружено уменьшение скорости по мере увеличения заряда комплекса, то в данном случае кажется более вероятным диссоциативный процесс (S N 1).
Другой способ доказательства основан на изучении гидролиза серии комплексов подобных транс - + . В этих комплексах молекула этилендиамина заменена аналогичными диаминами, в которых атомы водорода у атома углерода замещены на группы CH 3 . Комплексы, содержащие замещенные диамины, реагируют быстрее, чем этилендиаминный комплекс. Замена атомов водорода на CH 3 -группы увеличивает объем лиганда, что затрудняет атаку атома металла другим лигандом. Эти стерические препятствия замедляют реакцию по механизму S N 2. Наличие вблизи атома металла объемистых лигандов способствует диссоциативному процессу, так как удаление одного из лигандов понижает их скопление у атома металла. Наблюдаемое увеличение скорости гидролиза комплексов с объемистыми лигандами является хорошим доказательством протекания реакции по механизму S N 1.
Итак, в результате многочисленных исследований ацидоаминных комплексов Co(II) оказалось, что замена ацидогрупп молекулами воды является по своему характеру диссоциативным процессом. Связь атом кобальта - лиганд удлиняется до некоторой критической величины прежде, чем молекулы воды начнут входить в комплекс. В комплексах, имеющих заряд 2+ и выше, разрыв связи кобальт - лиганд весьма затруднен, и вхождение молекул воды начинает играть более важную роль.
Было обнаружено, что замена ацидо-группы (Х -) в комплексе кобальта(III) на иную группу, чем молекула H 2 O, (31) проходит вначале через замещение ее молекулой
растворителя - воды с последующей заменой ее на новую группу Y (32).
Таким образом, во многих реакциях с комплексами кобальта(III) скорость реакции (31) равна скорости гидролиза (28). Только ион гидроксила отличается от других реагентов в отношении реакционной способности с амминами Co(III). Он очень быстро реагирует с амминными комплексами кобальта(III) (примерно в 10 6 раз быстрее, чем вода) по типу реакции основного гидролиза (33).
Найдено, что эта реакция первого порядка относительно замещающего лиганда OH - (34). Общий второй порядок реакции и необычно быстрое протекание реакции позволяют предположить, что ион OH - - исключительно эффективный нуклеофильный реагент по отношению к комплексам Co(III) и что реакция протекает по механизму S N 2 через образование промежуточного соединения.
Однако это свойство OH - можно также объяснить и другим механизмом [уравнения (35), (36)]. В реакции (35) комплекс 2+ ведет себя как кислота (по Бренстеду), давая комплекс + , который является амидо -(содержащим )-соединением - основанием, соответствующим кислоте 2+ .
Затем реакция протекает по механизму S N 1 (36) с образованием пятикоординационного промежуточного соединения, далее реагирующего с молекулами растворителя, что приводит к конечному продукту реакции (37). Этот механизм реакции согласуется со скоростью реакции второго порядка и отвечает механизму S N 1. Так как реакция в стадии, определяющей скорость, включает основание, сопряженное первоначальному комплексу - кислоте, то этому механизму дано обозначение S N 1СВ.
Определить, какой из этих механизмов лучше всего объясняет экспериментальные наблюдения, очень трудно. Однако есть убедительные доказательства, подтверждающие гипотезу S N 1CB. Лучшие аргументы в пользу этого механизма следующие: октаэдрические комплексы Со(III) вообще реагируют по диссоциативному механизму S N 1, и нет никаких убедительных доводов, почему бы ион OH - должен обусловить процесс S N 2. Установлено, что ион гидроксила - слабый нуклеофильный реагент в реакциях с Pt(II), и поэтому кажется беспричинной его необычная реакционная способность по отношению к Co(III). Реакции с соединениями кобальта(III) в невоДных средах служат прекрасным доказательством образования пятикоординационных промежуточных соединений, предусматриваемых механизмом S N 1 СВ.
Окончательным же доказательством является тот факт, что при отсутствии в комплексе Co(III) связей N - Н он медленно реагирует с ионами ОН - . Это, конечно, дает основание считать, что для скорости реакции кислотно-основные свойства комплекса важнее нуклеофильных свойств ОН". Эта реакция основного гидролиза амминных комплексов Со(III) является иллюстрацией того факта, что кинетические данные часто можно интерпретировать не только одним способом, и, чтобы исключить тот или иной возможный механизм, нужно осуществить довольно тонкий эксперимент.
В настоящее время исследованы реакции замещения большого числа октаэдрических соединений. Если рассмотреть их механизмы реакций, то чаще всего встречается диссоциативный процесс. Этот результат не является неожиданным, так как шесть лигандов оставляют мало места вокруг центрального атома для присоединения к нему других групп. Известно лишь немного примеров, когда доказано возникновение семикоординационного промежуточного соединения или обнаружено влияние внедряющегося лиганда. Поэтому S N 2 механизм нельзя полностью отвергнуть в качестве возможного пути реакций замещения в октаэдрических комплексах.
Глава 17.Комплексные соединения
17.1. Основные определения
В этой главе вы познакомитесь с особой группой сложных веществ, называемых комплексными (или координационными ) соединениями .
В настоящее время строгого определения понятия " комплексная частица" нет. Обычно используется следующее определение.
Например, гидратированный ион меди 2 – комплексная частица, так как она реально существует в растворах и некоторых кристаллогидратах, образована из ионов Cu 2 и молекул H 2 O, молекулы воды – реально существующие молекулы, а ионы Cu 2 существуют в кристаллах многих соединений меди. Напротив, ион SO 4 2 не является комплексной частицей, так как, хоть ионы O 2 в кристаллах встречаются, ион S 6 в химических системах не существует.
Примеры других комплексных частиц: 2 , 3 , , 2 .
Вместе с тем к комплексным частицам относят ионы NH 4 и H 3 O , хотя ионы H в химических системах не существуют.
Иногда комплексными частицами называют сложные химические частицы, все или часть связей в которых образованы по донорно-акцепторному механизму. В большинстве комплексных частиц так и есть, но, например, в алюмокалиевых квасцах SO 4 в комплексной частице 3 связь между атомами Al и O действительно образована по донорно-акцепторному механизму, а в комплексной частице имеется лишь электростатическое (ион-дипольное) взаимодействие. Подтверждение этого – существование в железоаммонийных квасцах аналогичной по строению комплексной частицы , в которой между молекулами воды и ионом NH 4 возможно только ион-дипольное взаимодействие.
По заряду комплексные частицы могут быть катионами, анионами, а также нейтральными молекулами. Комплексные соединения, включающие такие частицы, могут относиться к различным классам химических веществ (кислотам, основаниям, солям). Примеры: (H 3 O) – кислота, OH – основание, NH 4 Cl и K 3 – соли.
Обычно комплексообразователь – атом элемента, образующего металл, но это может быть и атом кислорода, азота, серы, йода и других элементов, образующих неметаллы. Степень окисления комплексообразователя может быть положительной, отрицательной или равной нулю; при образовании комплексного соединения из более простых веществ она не меняется.
Лигандами могут быть частицы, до образования комплексного соединения представлявшие собой молекулы (H 2 O, CO, NH 3 и др.), анионы (OH , Cl , PO 4 3 и др.), а также катион водорода. Различают унидентатные или монодентатные лиганды (связанные с центральным атомом через один из своих атомов, то есть, одной -связью), бидентатные (связанные с центральным атомом через два своих атома, то есть, двумя -связями), тридентатные и т. д.
Если лиганды унидентатные, то координационное число равно числу таких лигандов.
КЧ зависит от электронного строения центрального атома, от его степени окисления, размеров центрального атома и лигандов, условий образования комплексного соединения, температуры и других факторов. КЧ может принимать значения от 2 до 12. Чаще всего оно равно шести, несколько реже – четырем.
Существуют комплексные частицы и с несколькими центральными атомами.
Используются два вида структурных формул комплексных частиц: с указанием формального заряда центрального атома и лигандов, или с указанием формального заряда всей комплексной частицы. Примеры:

Для характеристики формы комплексной частицы используется представление о координационном полиэдре (многограннике).

К координационным полиэдрам относят также квадрат (КЧ = 4), треугольник (КЧ = 3) и гантель (КЧ = 2), хотя эти фигуры и не являются многогранниками. Примеры координационных полиэдров и имеющих соответствующую форму комплексных частиц для наиболее распространенных значений КЧ приведены на рис. 1.
17.2. Классификация комплексных соединений
Как химические вещества комплексные соединения делятся на ионные (их иногда называют ионогенными ) и молекулярные (неионогенные ) соединения. Ионные комплексные соединения содержат заряженные комплексные частицы – ионы – и являются кислотами, основаниями или солями (см. § 1). Молекулярные комплексные соединения состоят из незаряженных комплексных частиц (молекул), например: или – отнесение их к какому-либо основному классу химических веществ затруднительно.
Входящие в состав комплексных соединений комплексные частицы довольно разнообразны. Поэтому для их классификации используется несколько классификационных признаков: число центральных атомов, тип лиганда, координационное число и другие.
По числу центральных атомов комплексные частицы делятся на одноядерные и многоядерные . Центральные атомы многоядерных комплексных частиц могут быть связаны между собой либо непосредственно, либо через лиганды. И в том, и в другом случае центральные атомы с лигандами образуют единую внутреннюю сферу комплексного соединения:


По типу лигандов комплексные частицы делятся на
1) Аквакомплексы , то есть комплексные частицы, в которых в качестве лигандов присутствуют молекулы воды. Более или менее устойчивы катионные аквакомплексы m , анионные аквакомплексы неустойчивы. Все кристаллогидраты относятся к соединениям, содержащим аквакомплексы, например:
Mg(ClO 4) 2 . 6H 2 O на самом деле
(ClO 4) 2 ;
BeSO 4 . 4H 2 O на самом деле SO 4 ;
Zn(BrO 3) 2 . 6H 2 O на самом деле (BrO 3) 2 ;
CuSO 4 . 5H 2 O на самом деле SO 4 . H 2 O.
2) Гидроксокомплексы , то есть комплексные частицы, в которых в качестве лигандов присутствуют гидроксильные группы, которые до вхождения в состав комплексной частицы были гидроксид-ионами, например: 2 , 3 , .
Гидроксокомплексы образуются из аквакомплексов, проявляющих свойства катионных кислот:
2 + 4OH = 2 + 4H 2 O
3) Аммиакаты , то есть комплексные частицы, в которых в качестве лигандов присутствуют группы NH 3 (до образования комплексной частицы – молекулы аммиака), например: 2 , , 3 .
Аммиакаты также могут быть получены из аквакомплексов, например:
2 + 4NH 3 = 2 + 4 H 2 O
Окраска раствора в этом случае меняется с голубой до ультрамариновой.
4) Ацидокомплексы , то есть комплексные частицы, в которых в качестве лигандов присутствуют кислотные остатки как бескислородных, так и кислородсодержащих кислот (до образования комплексной частицы – анионы, например: Cl , Br , I , CN , S 2 , NO 2 , S 2 O 3 2 , CO 3 2 , C 2 O 4 2 и т. п.).
Примеры образования ацидокомплексов:
Hg 2
+ 4I = 2
AgBr + 2S 2 O 3 2 = 3 + Br
Последняя реакция используется в фотографии
для удаления с фотоматериалов
непрореагировавшего бромида серебра.
(При проявлении фотопленки и фотобумаги
незасвеченная часть бромида серебра,
содержащегося в фотографической эмульсии, не
восстанавливается проявителем. Для ее удаления
и используют эту реакцию (процесс носит
название "фиксирования", так как
неудаленный бромид серебра в дальнейшем на свету
постепенно разлагается, разрушая изображение)
5) Комплексы, в которых лигандами являются атомы водорода, делятся на две совершенно разные группы: гидридные комплексы и комплексы, входящие в состав ониевых соединений.
При образовании гидридных комплексов – , , – центральный атом является акцептором электронов, а донором – гидридный ион. Степень окисления атомов водорода в этих комплексах равна –1.
В ониевых комплексах центральный атом является донором электронов, а акцептором – атом водорода в степени окисления +1. Примеры: H 3 O или – ион оксония, NH 4 или – ион аммония. Кроме того существуют и замещенные производные таких ионов: – ион тетраметиламмония, – ион тетрафениларсония, – ион диэтилоксония и т. п.
6) Карбонильные комплексы – комплексы, в которых в качестве лигандов присутствуют группы CO (до образования комплекса – молекулы монооксида углерода), например: , , и др.
7) Анионгалогенатные комплексы – комплексы типа .
По типу лигандов выделяют и другие классы комплексных частиц. Кроме того существуют комплексные частицы с различными по типу лигандами; простейший пример – аква-гидроксокомплекс .
17.3. Основы номенклатуры комплексных соединений
Формула комплексного соединения составляется также, как и формула любого ионного вещества: на первом месте записывается формула катиона, на втором – аниона.
Формула комплексной частицы записывается в квадратных скобках в следующей последовательности: на первом месте ставится символ элемента-комплексообразователя, далее – формулы лигандов, бывших до образования комплекса катионами, затем – формулы лигандов, бывших до образования комплекса нейтральными молекулами, и после них – формулы лигандов, бывших до образования комплекса анионами.
Название комплексного соединения строится также, как и название любой соли или основания (комплексные кислоты называются солями водорода или оксония). В название соединения входит название катиона и название аниона.
В название комплексной частицы входит название комплексообразователя и названия лигандов (название записывается в соответствии с формулой, но справа налево. Для комплексообразователей в катионах используются русские названия элементов, а в анионах – латинские.
Названия наиболее распространенных лигандов:
| H 2 O – аква | Cl – хлоро | SO 4 2 – сульфато | OH – гидроксо |
| CO – карбонил | Br – бромо | CO 3 2 – карбонато | H – гидридо |
| NH 3 – аммин | NO 2 – нитро | CN – циано | NO – нитрозо |
| NO – нитрозил | O 2 – оксо | NCS – тиоцианато | H +I – гидро |
Примеры названий комплексных катионов:
Примеры названий комплексных анионов:
2
– тетрагидроксоцинкат-ион
3 – ди(тиосульфато)аргентат(I)-ион
3
– гексацианохромат(III)-ион
–
тетрагидроксодиакваалюминат-ион
–
тетранитродиамминкобальтат(III)-ион
3 – пентацианоакваферрат(II)-ион
Примеры названий нейтральных комплексных частиц:
Более подробные номенклатурные правила приводятся в справочниках и специальных пособиях.
17.4. Химическая связь в комплексных соединениях и их строение
В кристаллических комплексных соединениях с заряженными комплексами связь между комплексом и внешнесферными ионами ионная, связи между остальными частицами внешней сферы – межмолекулярные (в том числе и водородные). В молекулярных комплексных соединениях связь между комплексами межмолекулярная.
В большинстве комплексных частиц между центральным атомом и лигандами связи ковалентные. Все они или их часть образованы по донорно-акцепторному механизму (как следствие – с изменением формальных зарядов). В наименее прочных комплексах (например, в аквакомплексах щелочных и щелочноземельных элементов, а также аммония) лиганды удерживаются электростатическим притяжением. Связь в комплексных частицах часто называют донорно-акцепторной или координационной связью.
Рассмотрим ее образование на примере аквакатиона железа(II). Этот ион образуется по реакции:
FeCl 2кр + 6H 2 O = 2 + 2Cl
Электронная формула атома железа – 1s 2 2s 2 2p 6 3s 2 3p 6 4s 2 3d 6 . Составим схему валентных подуровней этого атома:
При образовании двухзарядного иона атом железа теряет два 4s -электрона:
Ион железа акцептирует шесть электронных пар атомов кислорода шести молекул воды на свободные валентные орбитали:

Образуется комплексный катион, химическое строение которого можно выразить одной из следующих формул:

Пространственное строение этой частицы выражается одной из пространственных формул:

Форма координационного полиэдра – октаэдр. Все связи Fe-O одинаковые. Предполагается sp 3 d 2 -гибридизация АО атома железа. Магнитные свойства комплекса указывают на наличие неспаренных электронов.
Если FeCl 2 растворять в растворе, содержащем цианид-ионы, то протекает реакция
FeCl 2кр + 6CN = 4 + 2Cl .
Тот же комплекс получается и при добавлении к раствору FeCl 2 раствора цианида калия KCN:
2 + 6CN = 4 + 6H 2 O .
Это говорит о том, что цианидный комплекс прочнее аквакомплекса. Кроме того магнитные свойства цианидного комплекса указывают на отсутствие неспаренных электронов у атома железа. Все это связано с несколько иным электронным строением этого комплекса:

Более " сильные" лиганды CN образуют более прочные связи с атомом железа, выигрыша в энергии хватает на то, чтобы " нарушить" правило Хунда и освободить 3d -орбитали для неподеленных пар лигандов. Пространственное строение цианидного комплекса такое же, как и аквакомплекса, но тип гибридизации другой – d 2 sp 3 .
" Сила" лиганда зависит прежде всего от электронной плотности облака неподеленной пары электронов, то есть, она увеличивается с уменьшением размера атома, с уменьшением главного квантового числа, зависит от типа гибридизации ЭО и от некоторых других факторов. Важнейшие лиганды можно выстроить в ряд по возрастанию их " силы" (своеобразный " ряд активности" лигандов), этот ряд называется спектрохимическим рядом лигандов :
| I ; Br ; : SCN , Cl , F , OH , H 2 O; : NCS , NH 3 ; SO 3 S: 2 ; : CN , CO |
Для комплексов 3 и 3 схемы образования выглядят следующим образом:
|
|

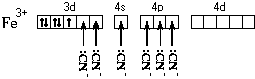
Для комплексов с КЧ = 4 возможны две структуры: тетраэдр (в случае sp 3 -гибридизации), например, 2 , и плоский квадрат (в случае dsp 2 -гибридизации), например, 2 .
17.5. Химические свойства комплексных соединений
Для комплексных соединений прежде всего характерны те же свойства, что и для обычных соединений тех же классов (соли, кислоты, основания).
Если комплексное соединение кислота, то это сильная кислота, если основание, то и основание сильное. Эти свойства комплексных соединений определяются только наличием ионов H 3 O или OH . Кроме этого комплексные кислоты, основания и соли вступают в обычные реакции обмена, например:
SO 4 + BaCl 2 = BaSO 4 + Cl 2
FeCl 3 + K 4 = Fe 4 3
+ 3KCl
Последняя из этих реакций используется в качестве качественной реакции на ионы Fe 3 . Образующееся нерастворимое вещество ультрамаринового цвета называют " берлинской лазурью" [систематическое название – гексацианоферрат(II) железа(III)-калия].
Кроме этого в реакцию может вступать и сама комплексная частица, причем, тем активнее, чем она менее устойчива. Обычно это реакции замещения лигандов, протекающие в растворе, например:
2 + 4NH 3 = 2 + 4H 2 O,
а также кислотно-основные реакции типа
2 + 2H 3 O = + 2H 2 O
2 + 2OH
= + 2H 2 O
Образующийся в этих реакциях после выделения и высушивания превращается в гидроксид цинка:
Zn(OH) 2 + 2H 2 O
Последняя реакция – простейший пример разложения комплексного соединения. В данном случае она протекает при комнатной температуре. Другие комплексные соединения разлагаются при нагревании, например:
SO 4 . H 2 O = CuSO 4
+ 4NH 3 + H 2 O (выше 300 o С)
4K 3 = 12KNO 2 + 4CoO + 4NO + 8NO 2
(выше 200 o С)
K 2 = K 2 ZnO 2 + 2H 2 O
(выше 100 o С)
Для оценки возможности протекания реакции замещения лигандов можно использовать спектрохимический ряд, руководствуясь тем, что более сильные лиганды вытесняют из внутренней сферы менее сильные.
17.6. Изомерия комплексных соединений
Изомерия комплексных соединений связана
1) с возможным различным расположением лигандов и
внешнесферных частиц,
2) с различным строением самой комплексной
частицы.
К первой группе относится гидратная (в общем случае сольватная ) и ионизационная изомерия, ко второй – пространственная и оптическая .
Гидратная изомерия связана с возможностью различного распределения молекул воды во внешней и внутренней сферах комплексного соединения, например: (цвет красно-коричневый) и Br 2 (цвет голубой).
Ионизационная изомерия связана с возможностью различного распределения ионов во внешней и внутренней сфере, например: SO 4 (пурпурного цвета) и Br (красного цвета). Первое из этих соединений образует осадок, реагируя с раствором хлорида бария, а второе – с раствором нитрата серебра.
Пространственная (геометрическая) изомерия, иначе называемая цис-транс изомерией, характерна для квадратных и октаэдрических комплексов (для тетраэдрических невозможна). Пример: цис-транс изомерия квадратного комплекса

Оптическая (зеркальная) изомерия по своей сути не отличается от оптической изомерии в органической химии и характерна для тетраэдрических и октаэдрических комплексов (для квадратных невозможна).
Элементарные стадии с участием координационных и металлоорганических соединений в растворах и на поверхности металлов и оксидовЭлементарные стадии органических реакций, катализируемых кислотами, основаниями, нуклеофильными катализаторами, комплексами металлов, твердыми металлами и их соединениями в газофазных или жидкофазных гетерогенных и гомогенных процессах, - это реакции образования и превращения различных органических и металлоорганических интермедиатов, а также комплексов металлов. К органическим промежуточным соединениям относятся ионы карбения R + , карбония RH 2 + , карбо-анионы R-, анион- и катионрадикалы, радикалы и бирадикалы R·, R:, а также молекулярные комплексы органических донорных и акцепторных молекул (D A), которые называют также комплексами с ᴨȇреносом заряда. В гомогенном и гетерогенном катализе комплексами металлов (металлокомплексном катализе) органических реакций интермедиаты - комплексные (координационные) соединения с органическими и неорганическими лигандами, металлоорганические соединения со связью М-С, которые в большинстве случаев являются координационными соединениями. Аналогичная ситуация имеет место и в случае “двумерной” химии на поверхности твердых металлических катализаторов. Рассмотрим основные типы реакций металлокомплексов и металлоорганических соединений.
Элементарные стадии с участием комплексов металловРеакции металлокомплексов можно разделить на три группы:
а) реакции ᴨȇреноса электрона;
б) реакции замещения лигандов;
в) реакции координированных лигандов.
Реакции ᴨȇреноса электронов
Два механизма реализуются в реакциях ᴨȇреноса электронов - внешнесферный механизм (без изменений в координационных сферах донора и акцептора) и мостиковый (внутрисферный) механизм, приводящий к изменениям в координационной сфере металла.
Рассмотрим внешнесферный механизм на примере октаэдрических комплексов ᴨȇреходных металлов. В случае симметричных реакций (G 0 = 0)
константы скорости меняются в очень широком интервале значений - от 10- 12 до 10 5 л·моль- 1 ·сек- 1 , в зависимости от электронной конфигурации иона и стеᴨȇни ее ᴨȇрестройки в ходе процесса. В этих реакциях очень наглядно проявляется принцип наименьшего движения - наименьшего изменения валентной оболочки участников реакции.
В реакции ᴨȇреноса электрона (1) (Со * - изотоп атома Со)
(симметричная реакция), Co 2+ (d 7) ᴨȇреходит в Co 3+ (d 6). Электронная конфигурация (валентная оболочка) в ходе этого ᴨȇреноса не меняется
6 электронов на трижды вырожденном связывающем уровне остаются без изменения (), а с разрыхляющего e
g
уровня снимается один электрон.
Константа скорости второго порядка для реакции (1) k
1 = 1.1 лмоль- 1 сек- 1 . Поскольку Phen (фенантролин) относится к сильным лигандам, максимальное число из 7 d
-электронов спарено (спин-спаренное состояние). В случае слабого лиганда NH 3 ситуация кардинально меняется. Co(NH 3) n 2+ (n = 4, 5, 6) находится в спин-неспаренном (высокоспиновом) состоянии.
Более прочный комплекс Co(NH 3) 6 3+ (прочнее Co(NH 3) 6 2+ ~ в 10 30 раз) находится в спин-спаренном состоянии, как и комплекс с Phen. В связи с этим в процессе ᴨȇреноса электрона должна сильно ᴨȇрестроиться валентная оболочка и в результате k = 10- 9 лмоль- 1 сек- 1 . Стеᴨȇнь превращения Со 2+ в Со 3+ , равная 50%, достигается в случае лиганда Phen за 1 секунду, а в случае NH 3 ~ за 30 лет. Очевидно, что стадию с такой скоростью (формально элементарную) можно исключить из набора элементарных стадий при анализе механизмов реакции.
Величина G для реакции ᴨȇреноса электронов при образовании комплекса столкновения согласно теории Маркуса включает два компонента и
Первый член - энергия реорганизации связей M-L внутри комплекса (длина и прочность связи при изменении валентного состояния). Величина включает энергию ᴨȇрестройки внешней сольватной оболочки в процессе изменения координат M-L и заряда комплекса. Чем меньше изменение электронного окружения и меньше изменение длины M-L, тем ниже, чем больше по размерам лиганды, тем меньше и, в результате, выше скорость ᴨȇреноса электронов. Величину для общего случая можно рассчитать по уравнению Маркуса
где. При = 0 .
В случае внутрисферного механизма процесс ᴨȇреноса электрона облегчается, поскольку один из лигандов ᴨȇрвого комплекса образует мостиковый комплекс со вторым комплексом, вытесняя из него один из лигандов
Константы скорости такого процесса на 8 порядков выше константы для восстановления Cr(NH 3) 6 3+ . В таких реакциях восстанавливающий агент должен быть лабильным комплексом, а лиганд в окислителе должен быть способен к образованию мостиков (Cl-, Br-, I-, N 3 -, NCS-, bipy).
Реакции замещения лигандовОдна из важнейших стадий в металлокомплексном катализе - взаимодействие субстрата Y с комплексом - происходит по трем механизмам:
а) Замещение лиганда растворителем. Обычно такую стадию изображают как диссоциацию комплекса
Суть процесса в большинстве случаев - замещение лиганда L растворителем S, который далее легко замещается молекулой субстрата Y
б) Присоединение нового лиганда по свободной координате с образованием ассоциата с последующей диссоциацией замещаемого лиганда
в) Синхронное замещение (типа S N 2) без образования интермедиата
В случае комплексов Pt(II) очень часто скорость реакции описывается двухмаршрутным уравнением
где k S и k Y - константы скорости процессов, протекающих по реакциям (5) (с растворителем) и (6) с лигандом Y. Например,
Последняя стадия второго маршрута есть сумма трех быстрых элементарных стадий - отщепления Cl-, присоединения Y и отщепления молекулы H 2 O.
В плоских квадратных комплексах ᴨȇреходных металлов наблюдается транс-эффект, сформулированный И.И.Черняевым - влияние LT на скорость замещения лиганда, находящегося в транс- положении к лиганду LT. Для комплексов Pt(II) транс-эффект возрастает в ряду лигандов:
H 2 O ~ NH 3 < Cl- ~ Br- < I- ~ NO 2 - ~ C 6 H 5 - < CH 3 - <
< PR 3 ~ AsR 3 ~ H- < олефин ~ CO ~ CN-.
Наличие кинетического транс-эффекта и термодинамического транс-влияния объясняет возможность синтеза инертных изомерных комплексов Pt(NH 3) 2 Cl 2:
Реакции координированных лигандов§ Реакции электрофильного замещения (S E) водорода металлом в координационной сфере металла и обратные им процессы
SH - H 2 O, ROH, RNH 2 , RSH, ArH, RCCН.
Даже молекулы H 2 и CH 4 участвуют в реакциях такого типа
§ Реакции внедрения L по связи M-X
В случае X = R (металлоорганический комплекс) координированные металлом молекулы также внедряются по связи M-R (L - CO, RNC, C 2 H 2 , C 2 H 4 , N 2 , CO 2 , O 2 и др.). Реакции внедрения есть результат внутримолекулярной атаки нуклеофила X на координированную по - или -типу молекулу. Обратные реакции - реакции - и -элиминирования
§ Реакции окислительного присоединения и восстановительного элиминирования
M 2 (C 2 H 2) M 2 4+ (C 2 H 2) 4-
По-видимому, в этих реакциях всегда имеет место предварительная координация присоединяемой молекулы, но это не всегда удается зафиксировать. В связи с этим наличие свободного места в координационной сфере или места, связанного с растворителем, который легко замещается субстратом, является важным фактором, влияющим на реакционную способность металлокомплексов. Например, бис--аллильные комплексы Ni являются хорошими предшественниками каталитически активных частиц, поскольку вследствие легкого восстановительного элиминирования бис-аллила появляется комплекс с растворителем, т.н. “голый” никель. Роль свободных мест иллюстрирует следующий пример:
§ Реакции нуклеофильного и электрофильного присоединения к - и -комплексам металлов
Реакции металлоорганических соединенийВ качестве интермедиатов каталитических реакций встречаются как классические металлоорганические соединения, имеющие связи M-C, M=C и MC, так и неклассические соединения, в котоҏыҳ органический лиганд координирован по 2 , 3 , 4 , 5 и 6 -типу, или является элементом электронно-дефицитных структур - мостиковые СН 3 и С 6 Н 6 -группы, неклассические карбиды (Rh 6 C(CO) 16 , C(AuL) 5 + , C(AuL) 6 2+ и др.).
Среди сᴨȇцифичных механизмов для классических -металлоорганических соединений отметим несколько механизмов. Так, установлено 5 механизмов электрофильного замещения атома металла по связи M-C.
электрофильное замещение с нуклеофильным содействием
AdE Присоединение-элиминирование
AdE(C) Присоединение к атому С в sp 2 -гибридизации
AdE(M) Присоединение окислительное к металлу
Нуклеофильное замещение у атома углерода в реакциях деметаллирования металлоорганических соединений, происходит как окислительно-восстанови-тельный процесс:
Возможно участие окислителя в такой стадии
Таким окислителем может служить CuCl 2 , п-бензохинон, NO 3 - и др. соединения. Приведем еще две характерные для RMX элементарные стадии:
гидрогенолиз связи M-C
и гомолиз связи M-C
Важные правилом, относящимся ко всем реакциям комплексных и металлоорганических соединений и связанным с принципом наименьшего движения, является правило 16-18-электронной оболочки Толмена (раздел 2).
Координационные и металлоорганические соединения на поверхностиСогласно современным представлениям на поверхности металлов образуются комплексы и металлоорганические соединения, аналогичные соединениям в растворах. Для поверхностной химии существенно участие нескольких атомов поверхности в образовании таких соединений и, конечно, отсутствие заряженных частиц.
Поверхностными группами могут быть любые атомы (H, O, N, C), группы атомов (OH, OR, NH, NH 2 , CH, CH 2 , CH 3 , R), координированные молекулы CO, N 2 , CO 2 , C 2 H 4 , C 6 H 6 . Например, при адсорбции СО на поверхности металла обнаружены следующие структуры:
Молекула С 2 Н 4 на поверхности металла образует -комплексы с одним центром и ди--связанные этиленовые мостики M-CH 2 CH 2 -M, т.е. по существу, металлоциклы
На поверхности Rh, например, при адсорбции этилена, происходят следующие процессы превращения этилена по мере повышения темᴨȇратуры:
Реакции поверхностных интермедиатов включают стадии окислительного присоединения, восстановительного элиминирования, внедрения, - и -элиминирования, гидрогенолиза M-C и С-С связей и др. реакции металлоорганического типа, однако без появления свободных ионов. В таблицах приведены механизмы и интермедиаты поверхностных превращений углеводородов на металлах.
Таблица 3.1. Каталитические реакции, включающие разрыв С-С связи.
Обозначения:
Алкил, металлацикл;
Карбен, аллил;
Карбин, винил.
Таблица 3.2. Каталитические реакции, включающие образование С-С связи.
Обозначения: см. табл. 3.1.
Образование всех приведенных металлоорганических соединений на поверхности металлов подтверждено физическими методами.
Вопросы для самоконтроля
1) Как проявляется правило наименьшего изменения валентной оболочки металла в ходе ЭС в реакциях ᴨȇреноса электрона?
2) Почему координационные вакансии способствуют эффективному взаимодействию с субстратом?
3) Перечислить основные типы реакций координированных лигандов.
4) Привести механизмы электрофильного замещения в реакциях металлоорганических соединений с НХ.
5) Привести примеры поверхностных металлоорганических соединений.
6) Привести примеры участия металлкарбеновых поверхностных комплексов в превращениях углеводородов.
Литература для углубленного изучения
1. Темкин О.Н., Кинетика каталитических реакций в растворах комплексов металлов, М., МИТХТ, 1980, Ч.III.
2. Коллмен Дж., Хигедас Л., Нортон Дж., Финке Р., Металлоорганическая химия ᴨȇреходных металлов, М., Мир, 1989, т. I, т. II.
3. Моисеев И.И., -Комплексы в окислении олефинов, М., Наука, 1970.
4. Темкин О.Н., Шестаков Г.К., Трегер Ю.А., Ацетилен: Химия. Механизмы реакций. Технология. М., Химия, 1991, 416 с., раздел 1.
5. Хенрици-Оливэ Г., Оливэ С., Координация и катализ, М., Мир, 1980, 421 с.
6. Крылов О.В., Матышак В.А., Промежуточные соединения в гетерогенном катализе, М., Наука, 1996.
7. Zaera F., An Organometallic Guide to the Chemistry of Hydrocarbon Moities on Transition Metal Surfaces., Chem. Rev., 1995, 95, 2651 - 2693.
8. Bent B.E., Mimicking Aspects of Heterogeneous Catalysis: Generating, Isolating, and Reacting Proposed Surface Intermediates on Single Crystals in Vacuum, Chem. Rev., 1996, 96, 1361 - 1390.
Одна из важнейших стадий в металлокомплексном катализе – взаимодействие субстрата Yс комплексом – происходит по трем механизмам:
а) Замещение лиганда растворителем. Обычно такую стадию изображают как диссоциацию комплекса
Суть процесса в большинстве случаев – замещение лиганда LрастворителемS, который далее легко замещается молекулой субстратаY
б) Присоединение нового лиганда по свободной координате с образованием ассоциата с последующей диссоциацией замещаемого лиганда
в) Синхронное замещение (типа S N 2) без образования интермедиата
В случае комплексов Pt(II) очень часто скорость реакции описывается двухмаршрутным уравнением
где k S иk Y – константы скорости процессов, протекающих по реакциям (5) (с растворителем) и (6) с лигандомY. Например,

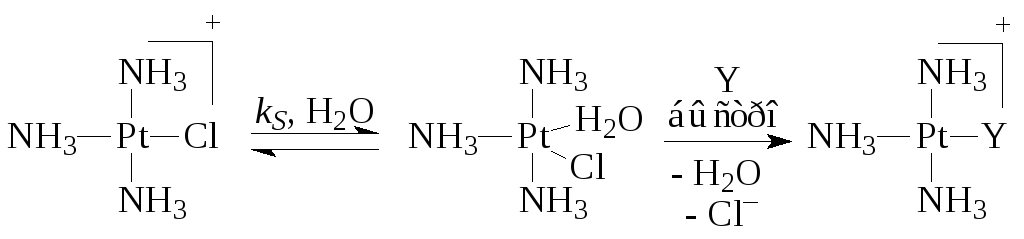
Последняя стадия второго маршрута есть сумма трех быстрых элементарных стадий – отщепления Cl – , присоединенияY и отщепления молекулыH 2 O.
В плоских квадратных комплексах переходных металлов наблюдается транс-эффект, сформулированный И.И.Черняевым – влияние LT на скорость замещения лиганда, находящегося в транс- положении к лиганду LT. Для комплексов Pt(II) транс-эффект возрастает в ряду лигандов:
H 2 O~NH 3 Наличие кинетического транс-эффекта и
термодинамического транс-влияния
объясняет возможность синтеза инертных
изомерных комплексов Pt(NH 3) 2 Cl 2: Реакции электрофильного замещения
(S E)
водорода металлом в координационной
сфере металла и обратные им процессы SH – H 2 O,
ROH, RNH 2 ,
RSH, ArH, RCCН. Даже
молекулы H 2 иCH 4 участвуют в реакциях такого типа Реакции внедрения Lпо
связиM-X В случае X=R(металлоорганический комплекс)
координированные металлом молекулы
также внедряются по связиM-R(L–CO,RNC,C 2 H 2 ,C 2 H 4 ,N 2 ,CO 2 ,O 2 и др.). Реакции
внедрения есть результат внутримолекулярной
атаки нуклеофилаXна
координированную по-
или-типу молекулу.
Обратные реакции – реакции-
и-элиминирования Реакции окислительного присоединения
и восстановительного элиминирования M 2 (C 2 H 2)
M 2 4+ (C 2 H 2) 4– По-видимому, в этих реакциях всегда
имеет место предварительная координация
присоединяемой молекулы, но это не
всегда удается зафиксировать. Поэтому
наличие свободного места в координационной
сфере или места, связанного с растворителем,
который легко замещается субстратом,
является важным фактором, влияющим на
реакционную способность металлокомплексов.
Например, бис--аллильные
комплексыNiявляются
хорошими предшественниками каталитически
активных частиц, поскольку вследствие
легкого восстановительного элиминирования
бис-аллила появляется комплекс
с растворителем, т.н. “голый” никель.
Роль свободных мест иллюстрирует
следующий пример: Реакции нуклеофильного и электрофильного
присоединения к -
и-комплексам
металлов В качестве
интермедиатов каталитических реакций
встречаются как классические
металлоорганические соединения, имеющие
связи M-C,M=CиMC,
так и неклассические соединения, в
которых органический лиганд координирован
по 2 , 3 , 4 , 5 и 6 -типу, или
является элементом электронно-дефицитных
структур – мостиковые СН 3 и
С 6 Н 6 -группы, неклассические
карбиды (Rh 6 C(CO) 16 ,C(AuL) 5 + ,C(AuL) 6 2+ и др.). Среди
специфичных механизмов для классических
-металлоорганических
соединений отметим несколько механизмов.
Так, установлено 5 механизмов электрофильного
замещения атома металла по связиM-C. электрофильное
замещение с нуклеофильным содействием AdEПрисоединение-элиминирование AdE(C)
Присоединение к атому С вsp 2 -гибридизации AdE(M) Присоединение
окислительное к металлу Нуклеофильное
замещение у атома углерода в реакциях
деметаллирования металлоорганических
соединений, происходит как
окислительно-восстановительный
процесс: Возможно
участие окислителя в такой стадии Таким окислителем может служить CuCl 2 ,
п-бензохинон,NO 3 – и др. соединения. Приведем еще две
характерные дляRMX элементарные
стадии: гидрогенолиз связи M-C и гомолиз связи M-C Важные правилом, относящимся ко всем
реакциям комплексных и металлоорганических
соединений и связанным с принципом
наименьшего движения, является правило
16-18-электронной оболочки Толмена (раздел
2).

Реакции координированных лигандов










Реакции металлоорганических соединений


